Analiza in silico wpływu substytucji aminokwasów na termostabilność syntazy skrobiowej - praca badawcza z Olimpiady Biologicznej
Przedstawiam ciekawą pracę badawczą ukazująca nieco inne podejście do badań, z wykorzystaniem symulacji dynamiki molekularnej. Praca została zgłoszona na Olimpiadzie Biologicznej i została oceniona na prawie maksymalną liczbę punktów (43/44 pkt). Jej recenzje załączyłem pod artykułem. Zachęcam do lektury.
Zamieszczone na stronie prace służą jedynie zapoznaniu się z ogólnym charakterem prac badawczych zgłaszanych na Olimpiadę Biologiczną. Niedopuszczalne jest kopiowanie całej koncepcji wykonania pracy bądź fragmentów jej tekstu we własnej pracy. Tego typu działania będą uznawane za plagiat i mogą wiązać się z odrzuceniem pracy przez komisję olimpiady.
Analiza in silico wpływu substytucji pojedynczych aminokwasów na termostabilność syntazy skrobiowej SSI jęczmienia zwyczajnego (Hordeum vulgare L.)
AUTOR: Olga Małgorzata Kowalska
Streszczenie
Zjawiska temperaturowe związane ze zmianami klimatu stanowią zagrożenie dla upraw jęczmienia (Hordeum vulgare L.). W wyniku długotrwałej ekspozycji na wysokie temperatury inaktywacji ulegają syntazy rozpuszczalnej skrobi, między innymi SSI, odpowiedzialna za pierwsze etapy syntezy tego wielocukru. Rozwiązaniem problemu może być substytucja wybranych aminokwasów tego białka, prowadząca do zwiększenia jego termostabilności. W pracy wykorzystano różne narzędzia bioinformatyczne, takie jak RosettaDesign, algorytmy oceniające energię swobodną polipeptydu oraz CABS-flex 2.0, przeprowadzający symulacje dynamiki molekularnej za pomocą modelu gruboziarnistego, aby przeprowadzić substytucje wybranych aminokwasów, cechujących się najwyższymi wartościami czynnika temperaturowego B, oraz zbadać in silico ich wpływ na termostabilność SSI. Zgodnie z analizami bioinformatycznymi, spośród przeprowadzonych substytucji najbardziej termostabilizujący wpływ wykazują E544R i K541N. Wymagane są dalsze badania in vitro, by zweryfikować ich wpływ na stabilność termiczną i aktywność enzymatyczną HvSSI.
Wstęp
Jęczmień (Hordeum vulgare L.) jest ważnym gatunkiem zbóż – większość jego upraw zlokalizowana jest w krajach rozwijających się, zamieszkiwanych przez ubogą ludność, z uwagi na możliwość wzrostu na dużych wysokościach, na terenach suchych, o dużym zasoleniu gleby (Grando i Macpherson, 2005). Globalny wzrost temperatur powodowany zmianami klimatu może stać się przyczyną znacznego obniżenia plonów jęczmienia – udowodniono, że długotrwała ekspozycja na podwyższone temperatury skutkuje obniżeniem masy ziaren i zawartości w nich skrobi (Wallwork i wsp., 1998). Jedną z przyczyn może być inaktywacja termolabilnych enzymów odpowiedzialnych za niektóre etapy syntezy skrobi, między innymi syntazy rozpuszczalnej skrobi I - SSI (Denyer i wsp., 1994).
SSI stanowi jedną z czterech klas syntaz rozpuszczalnej skrobi (I, II, III, IV), z których każda pełni inną rolę w syntezie tego węglowodanu z uwagi na właściwości fizykochemiczne i preferencje w wyborze substratu. Substratem SSI są przede wszystkim krótkie, proste łańcuchy glukozy, które wydłuża do średnich rozmiarów. Stanowi przy tym najaktywniejszą frakcję syntaz rozpuszczalnej skrobi w bielmie zbóż – np. w bielmie ryżu odpowiada za 70% całkowitej aktywności SS (Cuesta-Seijo i wsp., 2013).
Rozwiązaniem problemu mniej wydajnego gromadzenia skrobi w wysokich temperaturach może okazać się zaprojektowanie bardziej termostabilnej SSI. Projektowanie białek o zwiększonej termostabilności, za pomocą różnych strategii i narzędzi bioinformatycznych, odnajduje swoje zastosowanie w wielu dziedzinach nauki i przemysłu, gdzie kluczowe jest prowadzenie reakcji w temperaturach powyżej optimum termicznego danego enzymu (Modarres i wsp., 2016). Jedno z podejść mających na celu termostabilizację białka opiera się na wykorzystaniu uzyskiwanych z danych strukturalnych (pochodzących z rentgenografii strukturalnej) czynników Debye’a-Wallera (czynników temperaturowych B), opisujących termiczny nieporządek i oscylacje atomów. Regiony charakteryzujące się większymi wartościami czynnika temperaturowego B odznaczają się większą elastycznością (Sun i wsp., 2019) i z tego powodu mogą prowokować szybsze rozwinięcie się białka (Huang i wsp., 2017). Poprzez substytucję aminokwasu o wysokim czynniku Debye’a-Wallera do aminokwasu o większej „sztywności” udało się zwiększyć termostabilność kilku enzymów, takich jak lipaza B grzyba Candida antarctica (H. S., Kim i wsp., 2010). W przypadku syntaz skrobiowych jęczmienia zwyczajnego, w opisany powyżej sposób można zwiększyć termostabilność jedynie SSI, gdyż trójwymiarowe struktury pozostałych nie zostały jeszcze zdeponowane w bazach danych takich jak RCSB Protein Data Bank.
W pracy zbadano wpływ substytucji aminokwasów o największych wartościach czynnika temperaturowego B na termostabilność SSI z jęczmienia zwyczajnego (HvSSI). Aby móc ocenić zmiany w termostabilności w warunkach in silico, przenalizowano różnice w wartościach energii swobodnej różnych wariantów HvSSI, obliczone za pomocą różnych algorytmów, oraz przeprowadzono symulacje dynamiki wybranych mutantów za pomocą modelu gruboziarnistego.
Materiały i metody
Strukturę białka HvSSI oraz wartości czynników temperaturowych B poszczególnych aminokwasów pozyskano z bazy RCSB Protein Data Bank (PDB ID: 4HLN). Pobrana struktura nie jest kompletna – w porównaniu z oryginalną sekwencją brakuje w niej 94 aminokwasów od N-końca, a także dwóch pętli – od 116. do 123. oraz od 201. do 206. aminokwasu, z uwagi na ich nieuporządkowanie (Cuesta-Seijo i wsp., 2013). Brakujące pętle prawdopodobnie pełnią ważną rolę w procesie wiązania substratu, dlatego postanowiono domodelować je, wykorzystując serwer Robetta, umożliwiający otrzymanie struktury 3D danego białka, między innymi za pomocą metody modelowania komparatywnego, którą wykorzystano w pracy (D. E. Kim i wsp., 2004).
Do dalszej analizy wykorzystano model otrzymany za pomocą Robetty, składający się z 518 aminokwasów. Do wizualizacji trójwymiarowej struktury cząsteczki wykorzystano program UCSF ChimeraX (Goddard i wsp., 2018). Osobne badanie wpływu substytucji każdego aminokwasu na inny spośród 20 aminokwasów białkowych okazałoby się bardzo pracochłonne, dlatego zdecydowano się na bardziej racjonalne podejście, oparte na mutowaniu aminokwasów o najwyższych wartościach czynnika Debye’a-Wallera, wyłączając te znajdujące się na końcach łańcucha białkowego, za pomocą RosettaDesign (Liu i Kuhlman, 2006). Algorytm tego programu zastępował aminokwas na danej pozycji innym (z wyłączeniem cysteiny), który z największym prawdopodobieństwem zwiększał stabilność białka.
Mutacje określone przez RosettaDesign jako najbardziej stabilizujące wprowadzano do struktury otrzymanej z Robetty za pomocą FoldIt Standalone - programu łączącego pakiet modelowania molekularnego Rosetta z interaktywnym interfejsem graficznym (Kleffner i wsp., 2017). Po substytucji danego aminokwasu dokonywano automatycznego a dopasowania następnie odnotowywano pozycji 30 najbliższych energię swobodną reszt aminokwasowych cząsteczki obliczoną (funkcje w „wiggle” programie i „shake”), FoldIt za pomocą algorytmu Rosetty. Energia ta podawana jest w sztucznych jednostkach REU (Rosetta Energy Units).
Warianty białka o energii niższej niż białko typu dzikiego eksportowano do plików w formacie PDB, by porównać wartości ich energii swobodnej obliczone według innego algorytmu, wbudowanego w program Swiss-Pdb Viewer, częściowo opartego na polu siłowym GROMOS96 (Guex i Peitsch, 1997). Energię tę, obliczaną w jednostkach kJ/mol kalkulowano z uwzględnieniem wszystkich występujących w białku oddziaływań.
Oprócz tego, postanowiono ocenić wpływ wybranych mutacji na funkcjonalność białka za pomocą serwera PMut. Mutacje uznane przez algorytm za destrukcyjne oznaczane są jako „disease”, pozostałe określane są jako neutralne (López-Ferrando i wsp., 2017).
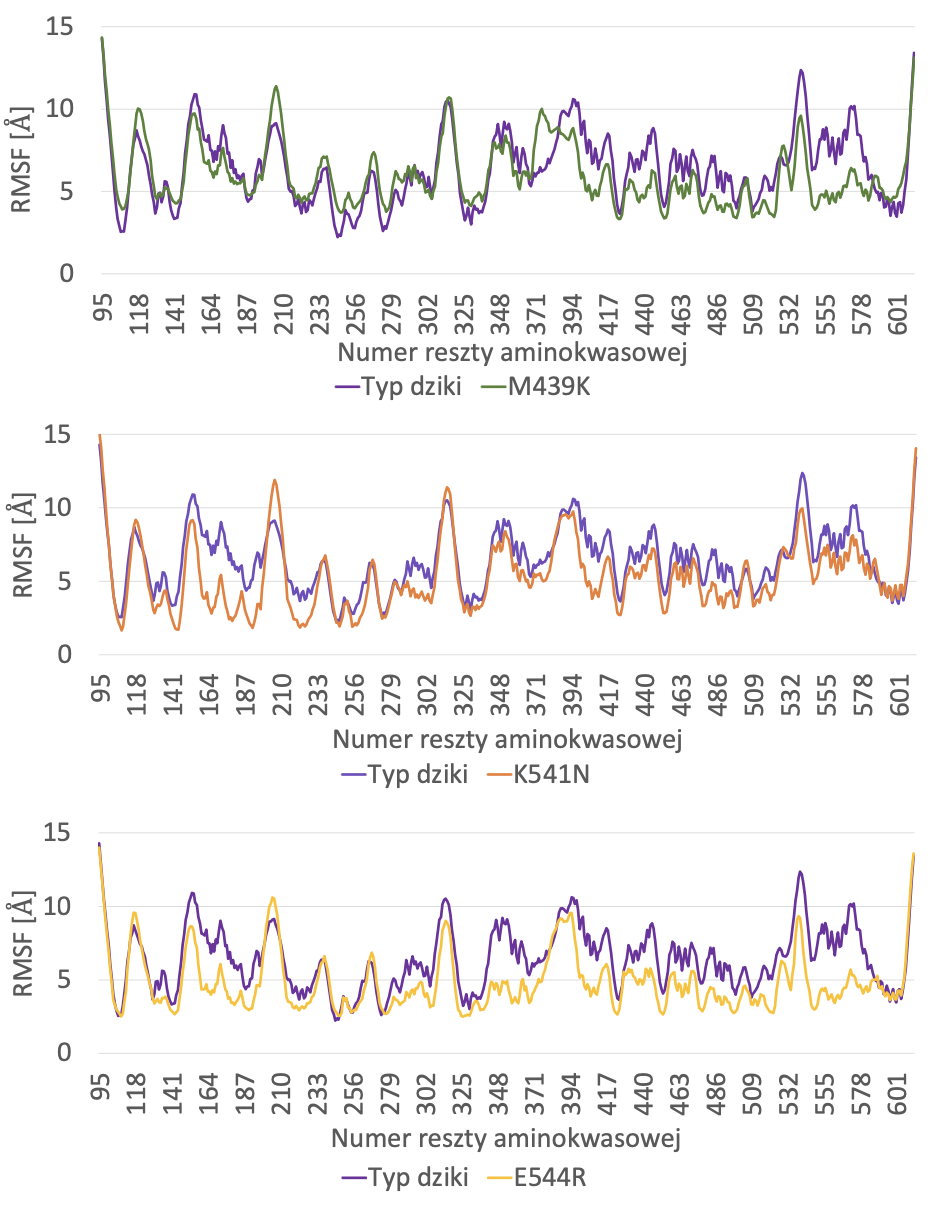
Do dalszej analizy wykorzystano mutanty odznaczające się ujemnymi wartościami zmian energii swobodnej według obu wykorzystywanych algorytmów i jednocześnie niewykazujące destrukcyjnego wpływu na funkcjonalność białka według obliczeń PMut. Wykonano symulacje elastyczności ich struktur w modelu gruboziarnistym na serwerze CABS-flex 2.0 (Kuriata i wsp., 2018). Zastosowano temperaturę symulacji T = 3, by wyraźniej unaocznić różnice między stabilnością poszczególnych wariantów. Pozostałe opcje symulacji pozostawiono w stanie domyślnym. Następnie porównano profile fluktuacji (RMSF, root mean square fluctuation – średnia kwadratów odchyleń położeń poszczególnych reszt aminokwasowych, obliczona na podstawie trajektorii CABS-flex- 2.0) wariantu niezmodyfikowanego oraz mutantów. Wartości RMSF pełnią podobną funkcję, jak czynniki temperaturowe B (Fuglebakk i wsp., 2012) – im są one wyższe, tym większą elastycznością charakteryzuje się dany region białka.
Wyniki
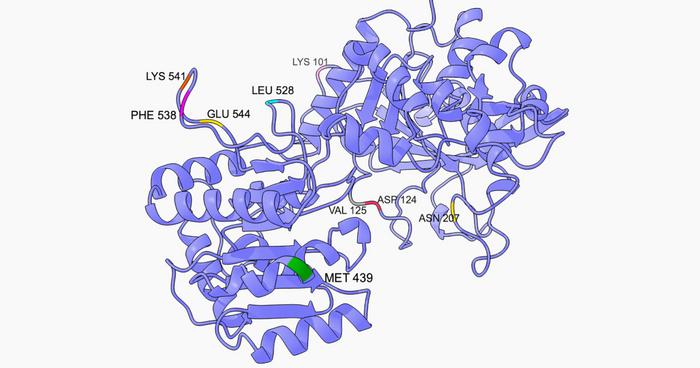
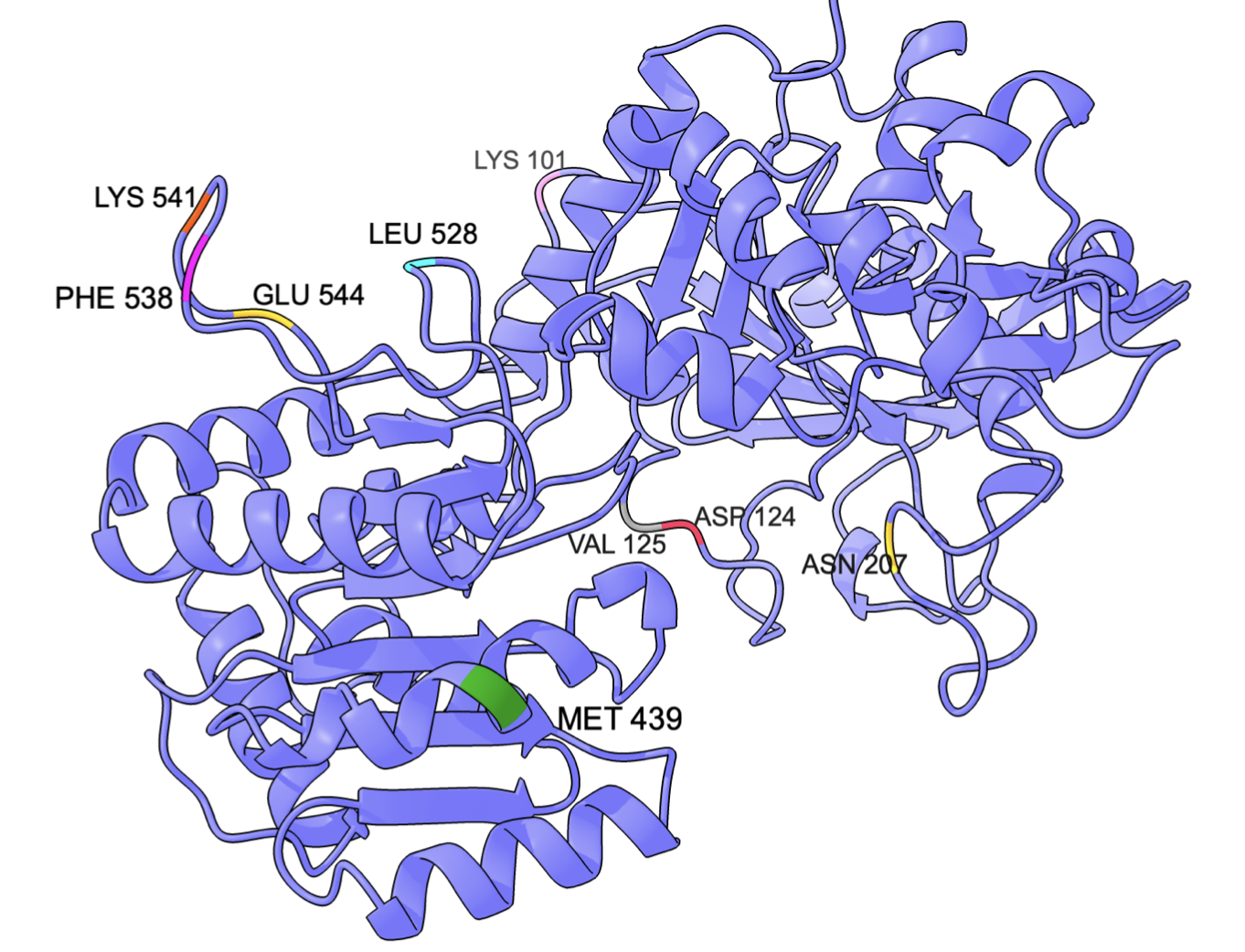
Za pomocą Robetty, w oparciu o strukturę zdeponowaną w bazie RCSB Protein Data Bank wygenerowano strukturę białka SSI jęczmienia zwyczajnego zawierającą aminokwasy 95-612. W dalszej pracy konsekwentnie stosowano tę numerację – pierwszy aminokwas wygenerowanej struktury oznaczano numerem 95 i tak dalej. Ostatecznie za pomocą RosettaDesign i FoldIt Standalone otrzymano 9 mutantów odznaczających się energią niższą od struktury pozyskanej z Robetty według algorytmu FoldIt Standalone: K101E, D124N, V125D, N207L, M439K, L528P, F538T, K541N, E544R. Lokalizacje 9 aminokwasów poddanych mutacjom przedstawiono na rycinie 1.
W tabeli (tab.1) porównano wartości energii swobodnej tych wariantów według obu wykorzystanych w pracy algorytmów a także wskazano, które z mutacji mogą upośledzać funkcjonalność białka według algorytmu PMut.

Do dalszych analiz wykorzystano tylko trzy mutanty: M439K, E544R, K541N. Pozostałe odrzucono ze względu na ich dodatnie wartości zmian energii swobodnej według obliczeń jednego z algorytmów lub prognozę wywoływania destrukcyjnego wpływu na strukturę białka według PMut, zakładając, że prawdopodobnie wykazują działanie destabilizujące lub upośledzają funkcje enzymu.
Porównano profile fluktuacji poddanych symulacjom wariantów (ryc. 2) z profilem fluktuacji białka niezmodyfikowanego. Spora część aminokwasów każdego wariantu charakteryzuje się mniejszymi wartościami RMSF od korespondujących z nimi aminokwasów białka dzikiego typu, jednak, szczególnie w przypadku M439K, niektóre z nich charakteryzują się wartościami wyższymi.
Dyskusja
Wykorzystanie narzędzi bioinformatycznych umożliwia znaczne usprawnienie badań w wielu dziedzinach przemysłu biotechnologicznego, między innymi w przemyśle rolniczym, umożliwiając modyfikowanie roślin uprawnych tak, by dawały lepsze plony oraz były bardziej odporne na szkodniki i zmiany klimatu (Ballabh i wsp.., 2017). Narzędzia pozwalające przewidzieć wpływ różnych modyfikacji na dane białko stanowią bardzo cenną pomoc w procesie projektowania między innymi enzymów o zwiększonej odporności na działanie wysokich temperatur, co zostało przedstawione w pracy.
Przeprowadzone analizy przewidywały potencjalnie destabilizujący wpływ większości wprowadzonych do białka substytucji. Tylko 3 z nich: M439K, E544R i K541N okazały się obniżać energię swobodną białka według obu zastosowanych algorytmów, nie wykazując przy tym destrukcyjnego wpływu na jego funkcjonalność według analiz PMut. Oznacza to, że właśnie te substytucje prawdopodobnie wykazywałyby działanie stabilizujące in vitro.
Substytucja M439K jako jedyna miała miejsce w alfa-helisie, a nie w luźniej zorganizowanym fragmencie białka. W tym przypadku metionina, aminokwas o niepolarnym łańcuchu, zamieniona została na lizynę z dodatnio naładowanym łańcuchem bocznym. Metionina 439 znajduje się w miejscu helisy wystawionym w stronę roztworu otaczającego białko, zatem zastąpienie jej aminokwasem polarnym, mogącym tworzyć wiązania wodorowe z cząsteczkami wody, może być czynnikiem stabilizującym (Vogt i Argos, 1997). Z drugiej strony, według analizy RMSF poszczególnych aminokwasów, substytucja ta może mieć jednak działanie destabilizujące – duża część aminokwasów charakteryzuje się wartościami RMSF równymi lub nawet wyższymi od korespondujących aminokwasów białka natywnego. Przyczyną może prawdopodobna być utrata hydrofobowych oddziaływań między metioniną a okolicznymi aminokwasami niepolarnymi.
Analizy przeprowadzone w programie CABS-Flex 2.0 wykazały, że prawdopodobnie najbardziej termostabilizujący wpływ na SSI mają mutacje E544R i K541N, gdyż wartości RMSF jedynie nielicznych aminokwasów tych mutantów były równe lub wyższe od korespondujących z nimi wartości RMSF białka typu dzikiego. Aminokwasy o pozycjach: 544 i 541 zlokalizowane są w pętli oddalonej od miejsca aktywnego enzymu, co daje nadzieję, że ich substytucje nie wpłyną negatywnie na katalizę.
Według analiz trójwymiarowych struktur białka, zamiana glutaminianu na argininę w pozycji 544 skutkuje wykształceniem nowego wiązania wodorowego między jej łańcuchem bocznym a szkieletem alaniny w pozycji 540, również wchodzącej w skład pętli, co może wyjaśniać stabilizujący wpływ substytucji E544R na SSI. Wiązania wodorowe uznawane są za jeden z najważniejszych czynników stabilizujących białka (Vogt i Argos, 1997). Wyjaśnienie potencjalnie stabilizującego wpływu substytucji K541N jest trudniejsze, gdyż w tym przypadku ani znajdująca się pierwotnie w tym miejscu lizyna, ani asparagina nie zdają się tworzyć żadnych dodatkowych wiązań czy oddziaływań z okolicznymi aminokwasami.
Główną zaletą wykorzystania modelu gruboziarnistego w przeprowadzeniu symulacji elastyczności struktur był krótszy czas oczekiwania na wyniki oraz mniejsze wymogi infrastrukturalne, niż w klasycznych symulacjach dynamiki molekularnej. Z drugiej strony, symulacje w modelu gruboziarnistym przeprowadzane są zbyt rzadko, by móc ostatecznie potwierdzić ich wiarygodność i skuteczność w badaniach nad termostabilnością (Modarres i wsp., 2016). Do dokładniejszych analiz potrzebna jest potężniejsza infrastruktura informatyczna, umożliwiająca przeprowadzenie pełnych symulacji dynamiki molekularnej, prowadzonych w skali nano- lub mikrosekund, które generują więcej przydatniejszych danych (Duan i Kollman, 1998).
Wraz z rozwojem technologii można spodziewać się dalszej poprawy wiarygodności analiz bioinformatycznych, nie są one jednak w stanie zastąpić badań in vitro i in vivo – aby potwierdzić termostabilizujący wpływ wprowadzonych substytucji, zwłaszcza najbardziej obiecujących E544R i K541N, a także sprawdzić ich wpływ na aktywność enzymatyczną HvSSI, należy przeprowadzić odpowiednie analizy doświadczalne.
Piśmiennictwo
- Ballabh, G., Ujjawal Kumar Singh Kushwaha, B., Deo, I., Prakash Jaiswal, J., i Prasad, B. (2017). Role of Bioinformatics in Crop Improvement. Global Journals Inc, 17.
- Cuesta-Seijo, J. A., Nielsen, M. M., Marri, L., Tanaka, H., Beeren, S. R., i Palcic, M. M. (2013). Structure of starch synthase i from barley: Insight into regulatory mechanisms of starch synthase activity. Acta Crystallographica Section D: Biological Crystallography , 69(6), 1013–1025.
- Denyer, K., Hylton, C., i Smith, A. (1994). The Effect of High Temperature on Starch Synthesis and the Activity of Starch Synthase. Functional Plant Biology, 21(6), 783.
- Duan, Y., i Kollman, P. A. (1998). Pathways to a protein folding intermediate observed in a 1- microsecond simulation in aqueous solution. Science , 282(5389), 740–744.
- Fuglebakk, E., Echave, J., i Reuter, N. (2012). Measuring and comparing structural fluctuation patterns in large protein datasets. Bioinformatics, 28(19), 2431–2440.
- Goddard, T. D., Huang, C. C., Meng, E. C., Pettersen, E. F., Couch, G. S., Morris, J. H., i Ferrin, T. E. (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Science, 27(1).
- Grando, S., i Macpherson, H. G. (2005). Food barley: importance, uses and local knowledge. Proceedings of the International Workshop on Food Barley Improvement, Hammamet, Tunisia, 14-17 January, 2002.
- Guex, N., i Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis, 18(15).
- Huang, J., Xie, D. F., i Feng, Y. (2017). Engineering thermostable (R)-selective amine transaminase from Aspergillus terreus through in silico design employing B-factor and folding free energy calculations. Biochemical and Biophysical Research Communications, 483(1).
- Kim, D. E., Chivian, D., i Baker, D. (2004). Protein structure prediction and analysis using the Robetta server. NucleicAcids Research , 32
- Kim, H. S., Le, Q. A. T., i Kim, Y. H. (2010). Development of thermostable lipase B from Candida antarctica (CalB) through in silico design employing B-factor and RosettaDesign. Enzyme and Microbial Technology, 47(1–2), 1–5.
- Kleffner, R., Flatten, J., Leaver-Fay, A., Baker, D., Siegel, J. B., Khatib, F., i Cooper, S. (2017). Foldit Standalone: a video game-derived protein structure manipulation interface using Rosetta. Bioinformatics , 33(17).
- Kuriata, A., Gierut, A. M., Oleniecki, T., Ciemny, M. P., Kolinski, A., Kurcinski, M., i Kmiecik, S. (2018). CABS-flex 2.0: A web server for fast simulations of flexibility of protein structures. Nucleic Acids Research, 46(W1).
- Liu, Y., i Kuhlman, B. (2006). RosettaDesign server for protein design. Nucleic Acids Research, 34.
- López-Ferrando, V., Gazzo, A., De La Cruz, X., Orozco, M., i Gelpí, J. L. (2017). PMut: A web- based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Research, 45(W1).
- Modarres, H. P., Mofrad, M. R., i Sanati-Nezhad, A. (2016). Protein thermostability engineering. RSCAdvances , 6 (116).
- Sun, Z., Liu, Q., Qu, G., Feng, Y., i Reetz, M. T. (2019). Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chemical Reviews.
- Vogt, G., i Argos, P. (1997). Protein thermal stability: Hydrogen bonds or internal packing? Folding and Design, 2(4), S40–S46.
- Wallwork, M. A. B., Logue, S. J., MacLeod, L. C., i Jenner, C. F. (1998). Effect of high temperature during grain filling on starch synthesis in the developing barley grain. Australian Journal of Plant Physiology, 25(2), 173–181.
- Zaloguj się albo zarejestruj aby dodać komentarz
