Folding@home - czyli jak wesprzeć naukowców nie wychodząc z domu

Obecnie coraz częściej w postępowaniu naukowym posługujemy się różnego rodzaju symulacjami. W niniejszym artykule przybliżę zastosowanie symulacji dynamiki molekularnej w dziedzinie biologii molekularnej i projektowaniu leków, przedstawię projekt Folding@home oraz wyjaśnię jak można dołożyć swoją cegiełkę do procesu poszukiwania leków, także w zakresie powstałego ostatnio zagrożenia w postaci COVID-19. Sam już dołączyłem się do opisywanego niżej programu.
Czym zajmuje się dynamika molekularna
Dynamika molekularna polega na przeprowadzeniu symulacji zachowania się atomów badanej cząsteczki, np. białka, w określonym środowisku. Zazwyczaj symulacje dotyczą środowiska wodnego, najbliższego właściwościami środowisku naturalnemu cząstek. Niestety symulacja faktycznego płynu fizjologicznego jest na ten moment zbyt skomplikowana. Dane wyjściowe do takiej symulacji uzyskuje się najczęściej eksperymentalnie, wprowadzając do symulacji początkowe dane m.in. dotyczące położenia atomów w cząsteczce i wiązań między nimi. Specjalne algorytmy wykorzystywane są do obliczenia krok po kroku zmian w kształcie cząsteczki. Uwzględniają one wiele zmiennych, takich jak rodzaje wiązań, siły i oddziaływania między atomami oraz ruch atomów w układzie. Dzięki temu możemy uzyskać model dynamicznej struktury złożonych cząstek. W przypadku białek jak wiemy struktura może być zmienna w zależności od tego jak dany łańcuch białkowy się sfałduje. Struktura ta może zmieniać się w czasie. Symulacja ma wymodelować podstawową strukturę przestrzenną białka jak i możliwe stany przejściowe.
Co daje nam takie modelowanie? Dzięki niemu możemy lepiej poznać strukturę przestrzenną kluczowych białek dla przebiegu wielu chorób (w tym zakaźnych). W szczególności kluczowe jest poznanie rejonów mogących być potencjalnym celem dla leków. Symulować można również proces łączenia się białek w kompleksy, np. receptor-ligand, czy przyłączenie leku do białka. Symulacja co prawda nie daje 100% pewności co do budowy i łączenia się białek, jednak może stanowić jeden z pierwszych etapów selekcji najbardziej obiecujących białek do dalszych, bardziej czasochłonnych i droższych badań. Dzięki temu można przyspieszyć skomplikowany proces opracowywania leku.
Folding@home - założenia projektu
Symulacje dynamiki molekularnej są bardzo złożone i wymagają olbrzymiej mocy obliczeniowej, aby czas ich wykonania mieścił się w sensownych granicach. Tego rzędu moce obliczeniowe posiadają najmocniejsze superkomputery, ale cóż, dostęp do nich jest mimo wszystko ograniczony. Naukowcy z Uniwersytetu Stanforda kilka lat temu wpadli na pomysł stworzenia projektu, umożliwiającego zebranie odpowiedniej mocy obliczeniowej. Wykorzystali do tego celu podłączone do sieci komputery wolontariuszy, którzy chcieli wesprzeć naukowców w badaniach nad lekami na wybrane choroby. Pojedynczy PC ma za małą mocz obliczeniową, ale setki tysięcy takich komputerów może zdziałać cuda. Dowodem na to może być ostatni odzew ludzi z całego świata na wieść, że Folding@home bierze udział w badaniu białek wirusa SARS-CoV-2. Liczba wolontariuszy przekroczyła 400 tys. a zebrana moc obliczeniowa osiągnęła zawrotne 470 petaFLOPS. Wiem, pewnie zrozumieliście tylko "zawrotne", ale żeby tą wartość nieco przybliżyć, można rzec, że jest to 2x tyle ile może na krótką metę wygenerować najpotężniejszy obecnie superkomputer. Na dłuższą metę, Folding@home osiągnął wydajność porównywalną do 7 najszybszych superkomputerów razem wziętych! Taka moc obliczeniowa może znacząco przyspieszyć prace nad białkami wirusa, które są w tym czasie objęte priorytetem.
Oprócz projektów związanych z COVID-19 Folding@home prowadzi symulacje dotyczące chorób nowotworowych (raka piersi, nerki, białka p53 oraz zmian epigenetycznych w nowotworach poprzez badanie struktury histonów), infekcyjnych (gorączki Denga, choroby Chagasa wywoływanej przez świdrowce, HCV - wirus zapalenia wątroby typu C, Ebola, wirusa Zika) oraz chorób związanych z nieprawidłowym fałdowaniem białek (choroba Alzheimera, Huntingtona i Parkinsona).
COVID-19 - Ty też możesz pomóc w znalezieniu leku!
Jak wspomniałem Folding@home jako priorytet wyznaczyło prace nad białkami wirusa SARS-CoV-2. Każdy posiadacz komputera stacjonarnego lub laptopa może przyłączyć się do tej batalii użyczając mocy obliczeniowej swojego sprzętu. Wystarczy zainstalować odpowiedni program (o czym niżej). Pewnie część z Was stwierdzi, że ma za słaby sprzęt, ale jak wspomniałem, liczy się to ile nas będzie.
Program umożliwia 3 stopniową regulację obciążenia komputera oraz udostępnianie mocy obliczeniowej w dowolnym momencie. Priorytet procesów związanych z obliczaniem symulacji ustawiony jest domyślnie na najniższym poziomie. Innymi słowy wszystko co robimy na komputerze będzie dla naszego procesora ważniejsze i wykonywane w pierwszej kolejności. Możemy np. udostępniać część mocy obliczeniowej w czasie mniej zasobożernych prac, bądź w czasie kiedy nie korzystamy z komputera. Niektórzy zostawiają komputer włączony na noc. Pomimo już uzyskanej ogromnej mocy obliczeniowej nowi wolontariusze nadal są potrzebni - obliczenia symulacji nawet przy takiej mocy trwają godzinami.
Mam nadzieję, że uda nam się jak najszybciej opracować skuteczny sposób walki z wirusem. Niemniej jednak liczę też na to, że nawet po zażegnaniu niebezpieczeństwa część osób zostanie w programie przyczyniając się być może do znalezienia leku na inną z badanych chorób. Sam przyłączyłem się do programu z myślą pozostania w nim na dłużej. Folding@home umożliwia tworzenie drużyn oraz zbieranie punktów w zależności od wykonanych obliczeń, co ma zmotywować wolontariuszy jako element przyjaznej rywalizacji. Stworzyliśmy także drużynę "BiologHelp Team" i liczę, że się do niej przyłączycie - ciekaw jestem ilu biolchemów podejmie wyzwanie :)
Jak przyłączyć się do programu
1. Pobierz program ze strony projektu: https://foldingathome.org/start-folding.
WAŻNE: Pamiętaj, aby program pobierać tylko z oficjalnej strony projektu. Ze względu na jego ostatnią popularność powstała kampania phishingowa, gdzie wysyłane są maile zachęcające do przyłączenia się do walki z koronawirusem i linkiem do złośliwej aplikacji.
2. Zainstaluj program. Postępuj zgodnie z instrukcjami i domyślnymi opcjami instalacji.


Pierwsze uruchomienie i konfiguracja programu
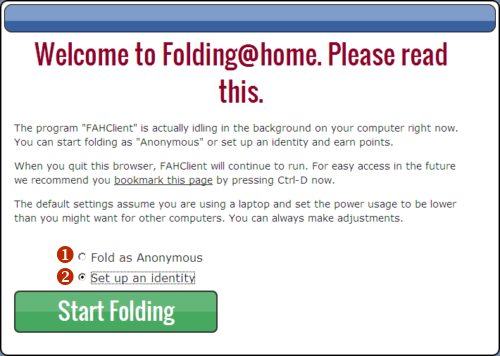

(1) - Pole wyboru nazwy użytkownika. Możemy tutaj wpisać cokolwiek. Jeżeli zostawimy pole puste, nie będzie do kogo przypisać punktów i takowe nie będą liczone
(2) - Pole wyboru drużyny. W tym polu można wpisać numer drużyny, do której chcecie dołączyć. Będzie mi miło, jeżeli dołączycie do BiologHelp Team wpisując numer: 255957 :). Po dołączeniu do drużyny i wsparciu pierwszej symulacji pojawicie się w statystykach jako członkowie tej drużyny, a Wasze punkty będą również wzbogacać konto danej drużyny (choć nie ma za to żadnych profitów oprócz ogromnej satysfakcji ;) Panel statystyk dostępny jest pod tym adresem: https://stats.foldingathome.org/team/255957 - tutaj akurat zobaczycie statystyki naszej drużyny, ale klikając na zakładki możecie znaleźć również ogólny ranking oraz inne drużyny czy użytkowników.
(3) - W tym polu możemy wpisać kod użytkownika, dzięki któremu zbierane punkty będą przypisane nie tylko drużynie, ale także użytkownikowi. Wymaga to podania nazwy użytkownika oraz adresu e-mail w formularzu na stronie https://apps.foldingathome.org/getpasskey. Po zatwierdzeniu formularza na podany adres e-mail przesłany zostanie kod użytkownika.
Żadne z powyższych pól nie jest obowiązkowe do wypełnienia aby rozpocząć obliczenia.
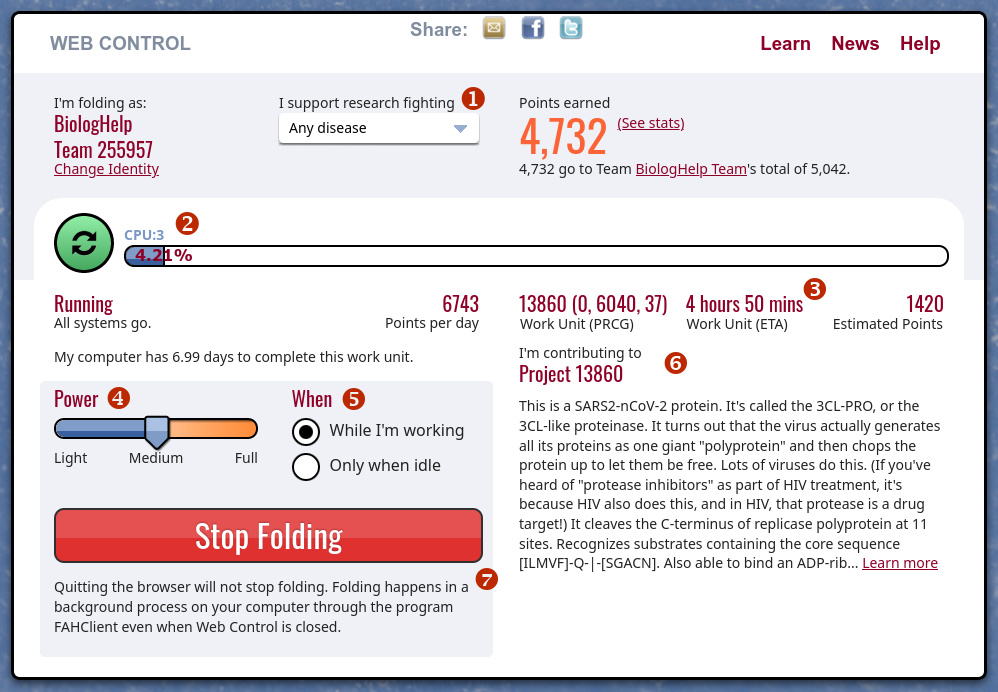
Po zatwierdzeniu nazwy użytkownika i ewentualnie wspomnianych dodatkowych opcji, pojawi się panel sterowania. Generalnie sam program działa w tle, natomiast jeżeli chcemy zobaczyć swoje postępy w uproszczonym panelu sterowania wystarczy, że wpiszemy w przeglądarkę adres https://client.foldingathome.org (można dodać do zakładek). Można też kliknąć prawym przyciskiem myszy na ikonce programu działającego w tle, na pasku zadań, i tam wybrać z listy "Web Control". Mogą wystąpić problemy z obsługą panelu w przeglądarce Chrome, dużo lepiej sprawdza się tutaj Firefox. Widoczne powyżej elementy panelu to:
(1) - Wybór grupy projektów, jakie chcemy wspierać. Aby wesprzeć badania białek wirusa SARS-CoV-2 należy pozostawić "Any disease" - jako że te badania mają obecnie ustalony najwyższy priorytet, nasza moc obliczeniowa zostanie automatycznie przekierowana do związanej z nimi symulacji, o ile takowa została ustawiona.
(2) - Pasek postępu obliczeń symulacji, przy której pomagamy. Dopiero po zakończeniu symulacji otrzymamy punkty.
(3) - Przewidywany czas do ukończenia symulacji.
(4) - Tutaj możemy wybrać jaką część mocy obliczeniowej komputera chcemy udostępnić, innymi słowy jak bardzo aplikacja obciąży komputer.
(5) - Tutaj ustawiamy czy chcemy, żeby obliczenia były wykonywane w tle podczas naszej pracy, czy też tylko w momencie kiedy nie korzystamy z komputera.
(6) - W tej części znajdziemy opis projektu / białka, do którego symulacji się przyczyniamy
(7) - w każdym momencie możemy zatrzymać obliczenia (np. kiedy uznamy, że za bardzo obciążają komputer, którego akurat potrzebujecie) klikając "Stop Folding". Warto pamiętać, że samo zamknięcie panelu sterowania / przeglądarki nie zatrzymuje obliczeń - te nadal prowadzone są w tle.
Dodatkowo program umożliwia podgląd animacji białka, nad którym aktualnie pracujemy. Wystarczy otworzyć Protein Viever w opcjach programu (prawy przycisk myszy na miniaturce w pasku zadań). Poniżej widok takiego podglądu (podobno można ustawić jako wygaszacz ekranu, ale jeszcze nie próbowałem).
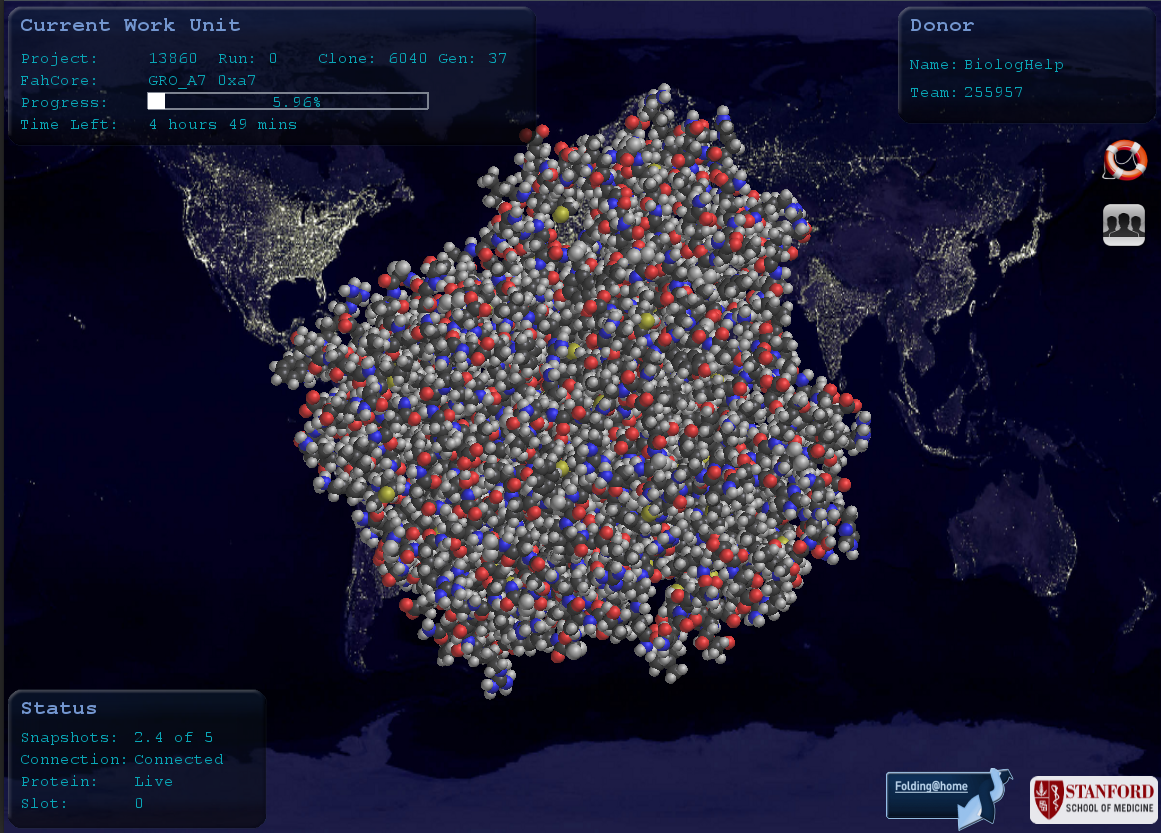
W dodatkowych opcjach znajdziecie także zaawansowane ustawienia programu (Advanced Control). Myślę, że Wam się nie przydadzą, ale w razie czego TUTAJ znajdziecie filmik opisujący jak zainstalować i skonfigurować program, również przez opcje zaawansowane.
Mam nadzieję, że zachęciłem Was do wsparcia wyżej przedstawionych działań :)
- Zaloguj się albo zarejestruj aby dodać komentarz
